About Us
Transforming Medicine Through the Science and Practice of Genetics and Genomics
The Department of Molecular and Human Genetics at Baylor College of Medicine is transforming medicine by expanding the role of genetics and genomics in science and medicine through major discoveries and integration of basic research, clinical, and diagnostic activities. Our talented faculty, trainees and staff have designed an environment that promotes and supports diversity, inclusion, and equity.
Our department consistently ranks first in the nation in funding from the National Institutes of Health. In addition to numerous NIH, National Science Foundation, and other competitive research grants, the faculty have received national recognition and support from the Pew Foundation and the Howard Hughes Medical Institute. Our educational programs attract the most highly qualified candidates. Our clinical genetics program, the largest in the country, offers patients unparalleled, single-source genetic testing and services.
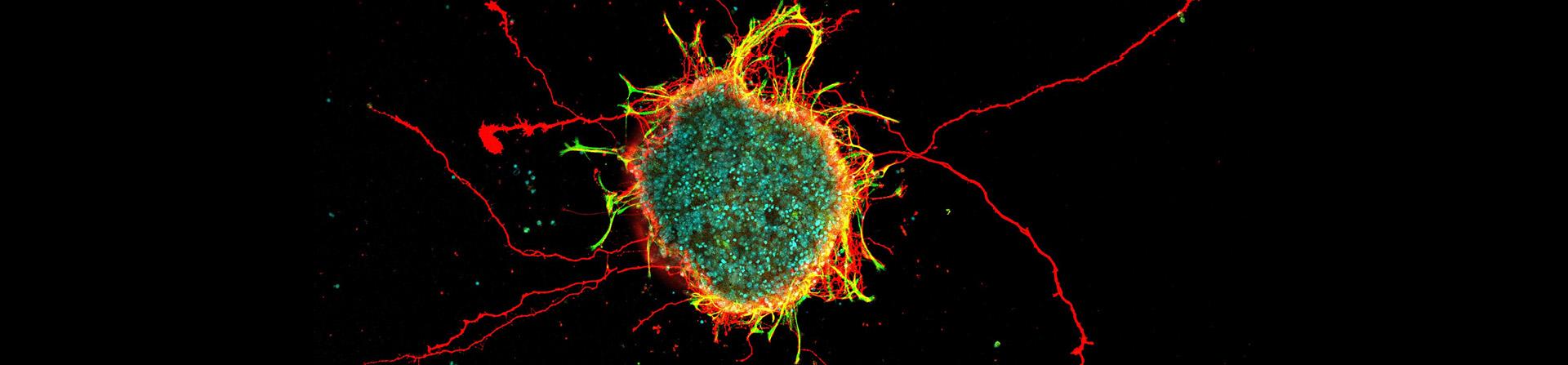
Featured Research Image (Above): This image showcases a neurosphere composed of retinal ganglion cells and glial cells derived from mouse retinas. The retinal ganglion cell axons are visualized in red, glial cells in green, and cell nuclei in cyan. Approximately 10,000 cells were aggregated and cultured in a U-bottom well to form this sphere, providing a robust model for studying axon outgrowth dynamics in retinal neurons. Image submitted by Han-Yin Jeng of Dr. Nicholas Tran's laboratory.

Message from the Chair
A message from our department chair, Brendan Lee, M.D., Ph.D.

Faculty
View listings of our department’s leadership and faculty with links to their bios.


Training Programs
We offer an array of valuable educational and training opportunities for medical students, graduate students, residents, fellows, and postdoctoral trainees who are interested in human and molecular genetics and genomics.

Basic & Clinical Genetics Research
Our department’s research covers such areas as genomics, DNA biology, gene expression, and the molecular basis of human disease. View our department’s clinical studies, research centers, projects and cores.

Clinical Services
Our clinical genetics program is the largest program of its kind in the country, with clinics spanning across multiple genetics-based disciplines.

Job Opportunities
Visit the BCM Career website and search the term "Molecular and Human Genetics" to find available positions within the Department of Molecular and Human Genetics.

Community Engagement
The Office of Community Engagement in the Department of Molecular and Human Genetics works to provide education and resources regarding genetics services to the community-at-large.

Giving
Give online or by mail to help support our department's research and community programs.

Diagnostic Laboratory
Baylor Genetics is dedicated to providing the medical genetics community with high-quality comprehensive diagnostic services.
Consultagene
Designed by the department, Consultagene is an online telehealth platform for genetics services.
