Infantile Spasms; Lissencephaly
This patient presented with repetitive spells of rapid flexion of the neck, extension of the arms, flexion of the hip and knee, and flexion of the abdominal muscles. These spells occurred in flurries, often upon awaking from sleep. The mother recognized them as unusual, but described them as excessive startle responses that were not provoked.
Other parents of children with this disorder describe their child as colicky and ascribe these spells to abdominal pain (the child seemingly flexes the abdominal musculature to guard). In children with colic (excessive crying in infancy), sudden flexion of the hips and knees may be seen. However, in colic there is not the extension of the arms nor the flexion of the neck described in this case. Also, the patient presented here did not have the excessive crying seen in colic.
A benign myoclonic condition with spells resembling this child's has been described in some infants. These children have normal development, a normal EEG and no other spells. Since the child presented here had visual inattentiveness (abnormal development) and hypsarrhythmia (abnormal EEG), he does not have this benign condition.
The spells described above combined with evidence of abnormal development (this is not always seen in this condition) and an abnormal EEG make infantile spasms the most likely diagnosis. These are seizures peculiar to infancy that usually present between 3-8 months of age. Infantile spasms are myoclonic seizures often accompanying severe brain malformations; they may occur in otherwise normal children. If accompanying brain disease, these seizures are termed symptomatic infantile spasms (85%). If no brain disease can be identified, these seizures are termed cryptogenic infantile spasms (15%). The prognosis and the response to treatment is poor in the symptomatic patients. Since brain disease, such as structural malformations, can accompany this disorder and since these brain malformations may impact upon the child more than the infantile spasms, it is prudent to perform neuroimaging in the child with infantile spasms.
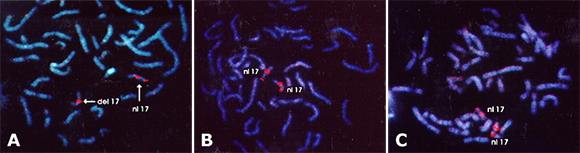
Patient #9 (Figure A), mother (Figure B), and father's (Figure C) chromosomes fluorescently labeled with a marker near the centromere for the 17th chromosomes delineates the 17th chromosomes with a bright orange label. Near the end of the short arm of the 17th chromosomes another marker for the region of the LIS-1 gene is shown with a fainter signal. The chromosomes delineated as "nl 17" is shown with LIS-1 probe properly labeling the LIS-1 gene. For mother (Figure B) and father (Figure C), the "del 17" chromosome does not show labeling with the LIS-1 probe, thus a deletion of genetic information in the region of the LIS-1 gene exists in this patient.
Both CT and MRI scans revealed a brain malformation consisting of a smooth brain (lissencephaly). It is important to properly identify brain malformations since the genetics vary with the type of brain malformation seen. In microcephaly, the brain may appear simple, but it is almost never smooth. This patient did not have microcephaly at birth, but has acquired it postnatally.
In pachygyria (part of the lissencephalic spectrum), there are areas of large simple gyri that may be near areas with normal gyrations. On CT scans, the brains of patients with pachygyria may appear smooth; MRI will usually distinguish pachygyria from lissencephaly. Polymicrogyria (many small gyrations), may give the appearance of a smooth brain but the MRI will usually distinguish these malformations from the lissencephalies.
So called cobblestone lissencephaly (named because of the pebbled appearance of the surface of the brain) accompanies Walker-Warburg syndrome and Fukyama Muscular Dystrophy. Hydrocephalus usually accompanies this form of lissencephaly.
This patient appears to have classic or type 1 lissencephaly such as that seen in Miller-Dieker lissencephaly (MDL), isolated lissencephaly sequence (ILS) or X-linked lissencephaly. These forms of lissencephaly are characterized by an extensive heterotopic zone of neurons that appear to have been arrested in the process of migration and the brain has a smooth cortical surface especially posteriorly. The brain is smooth owing to a lack of complexity of the outermost surface of the brain.
This patient had a deletion in the short arm of one 17th chromosome that is only detectable by fluorescent in situ hybridization (FISH) for the gene LIS-1. This gene deficit in one chromosome leads one to conclude that this patient has Miller-Dieker lissencephaly. Both the mother and father had normal FISH for LIS-1, therefore, they are unlikely to be carriers of a balanced translocation that could put them at risk for further affected children.
Lissencephaly (smooth brain) refers to the configuration of the cortical mantle in human disorders in which the cerebral cortex is severely malformed such that the surface of the cortex lacks the gyri and sulci seen in normal brain. In Miller-Dieker lissencephaly (MDL), the cortical malformation results from the arrest of migrating neurons in the formation of the cortical plate (Alvarez et al., 1986; Daube and Chou, 1966; Dieker et al., 1969; Miller, 1963). MDL and some cases of isolated lissencephaly sequence (ILS) result from the deletion of one copy of a gene on the short arm of the 17th chromosome, LIS-1 (Dobyns et al., 1993; Ledbetter et al., 1992; Reiner et al., 1993).
Patients with this brain malformation are severely mentally retarded, visually inattentive due to cortical blindness, and 80% have infantile spasms. Over 90% of patients with this disorder have seizures at some point in their life. Head circumference is typically normal at birth but a developing microcephaly is characteristic. This is due to the lack of complexity of the brain, improper synapse formation, and thus poor brain growth. A normal head size at birth is typical because the normal complement of neurons and cells are present. It is the location of these cells that is abnormal.
Miller-Dieker lissencephaly is a gene deletion syndrome. The minimal deletion necessary to result in lissencephaly involves one copy of the gene LIS-1. Most patients with isolated lissencephaly will have a deletion of this gene or they may have a deletion of another gene on the X chromosome as discussed below. This LIS-1 gene encodes a subunit of a brain platelet-activating factor acetylhydrolase, an enzyme that degrades platelet-activating factor. The deletion of one copy of this gene is all that is necessary to result in the lissencephaly phenotype.
Other genes close to the lissencephaly gene are probably necessary for some of the other characteristics of Miller-Dieker lissencephaly. Those other features include an upturned nares, micrognathia, a sacral dimple in 70% of patients, cardiac anomalies in 50% of patients, and deep palmer creases in 50 to 70% of patients. Somatic abnormalities are typically not seen in the isolated lissencephaly patients, thus Miller-Dieker lissencephaly is probably a contiguous gene deletion syndrome.
The LIS-1 gene encodes a protein that is a subunit of a brain platelet-activating factor (PAF) acetylhydrolase, an enzyme that degrades the lipid messenger, PAF. This enzyme complex is a novel serine esterase that is perhaps best classified as a calcium-independent phospholipase A2; it is pharmacologically related to brain acetylcholinesterase (Hattori et al., 1994a; Hattori et al., 1993; Hattori et al., 1994b). In addition, this form of brain PAF acetylhydrolase may serve as a signaling G-protein like complex (Ho et al., 1997). However, it is not known how LIS-1 functions in the development of the cortical plate, nor is it known how a hemideletion of LIS-1 and the resulting alteration in PAF degradation (or signaling) cause a neuronal migration disorder. It has been proposed that PAF serves as a neuronal cytoskeletal altering signal that can alter the process of neuronal migration (Clark et al., 1995).
Patients with lissencephaly are severely mentally retarded and require total supportive care. The lifespan of the patient with lissencephaly is not normal. These patients usually succumb to pulmonary infections owing to a lack of control of secretions.
At least one other classic lissencephaly locus exists. That locus appears to be on the X chromosome (Xq22.3) and only males have this form of lissencephaly. Female carriers of this gene have subcortical band heterotopia. The gene for this disorder has not been isolated. The brain pathology appears to be nearly identical to that of Miller-Dieker lissencephaly.
Infantile spasms are a myoclonic seizure type seen in infancy. The typical age of presentation is between three and eight months. Infantile spasms can either be symptomatic of an underlying neurologic or metabolic disorder, or can be cryptogenic which means that no underlying cause for the seizure can be found. The prognosis for infantile spasms in the symptomatic group is dismal. Prognosis in the cryptogenic group can be good, and some authors feel that early treatment with steroids, such as ACTH, improves the developmental outcome in these patients. This patient clearly had symptomatic infantile spasms. The EEG showed hypsarrhythmia (high voltage, disorganized, polyspikes) which is characteristic of infantile spasms. In this pattern of EEG, the recording of the electroencephalogram cannot be made at the standard amplitudes.
The seizures in lissencephaly are usually very difficult to control. The use of steroids (ACTH and prednisone) though typical for treatment of infantile spasms, may or may not be successful in the treatment of these seizures. In addition, seizures will return following treatment with steroids. Therefore, initiation of other anticonvulsants is appropriate in this condition.
Email comments: